CE-маркировка — регуляторные процессы Европейского Союза для медицинских изделий
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация — организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.

Скачать брошюру «CE-маркировка для медицинских изделий» в PDF-формате
Как зарегистрировать медицинское изделие в ЕС. Читать статью.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
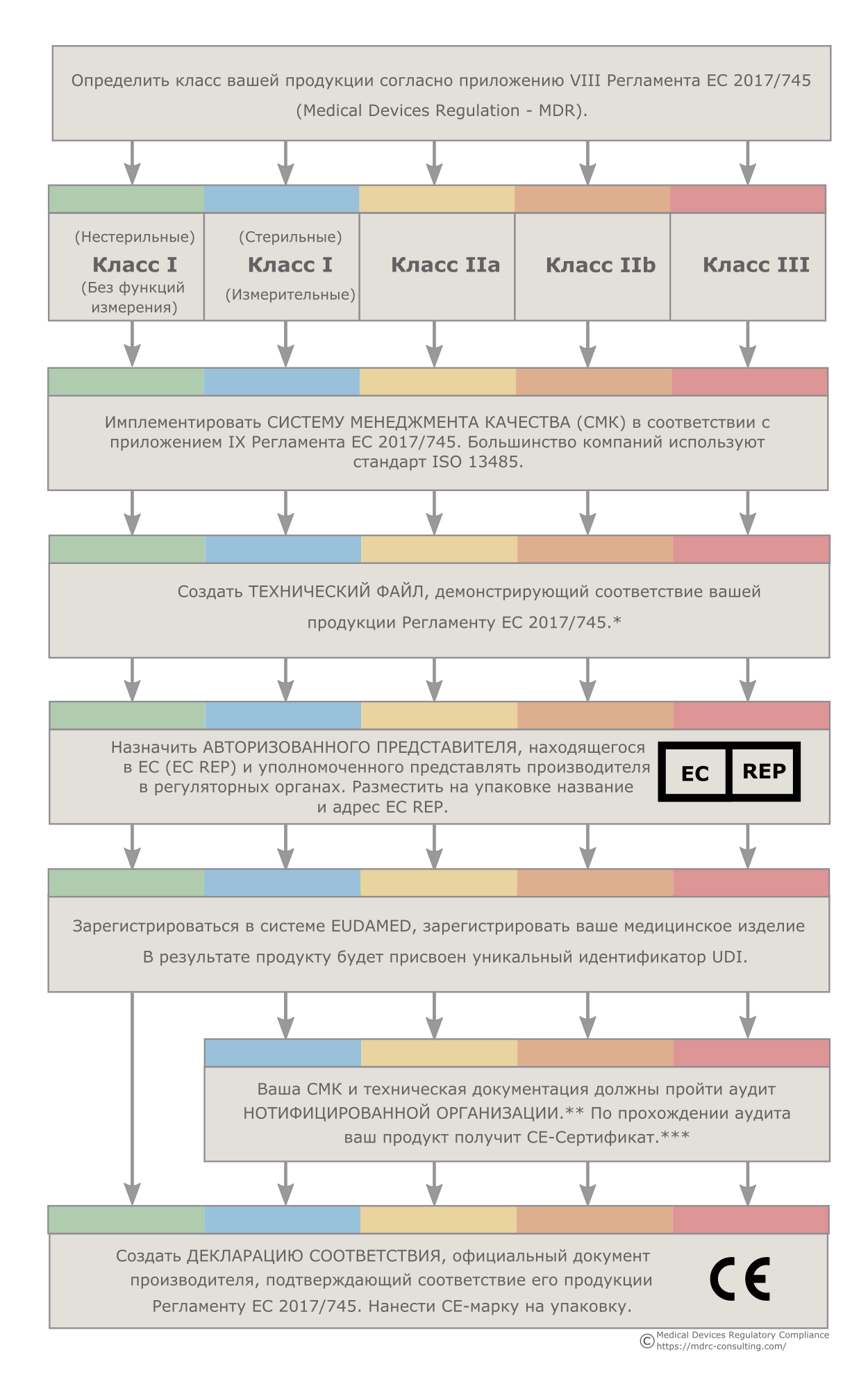
Классификация изделий медицинского назначения
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Обучение: CE-маркировка и регуляторная система ЕС для медицинских изделий
Технические файлы и досье разработки
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований — в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
ISO 13485 и система менеджмента качества (СМК)
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.
MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Авторизованное представительство в ЕС (EC REP)
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
Аудиты на соответствие требованиям Регламента ЕС 2017/745 и ISO 13485
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Взаимодействие с Нотифицированной Организацией
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Оценка и квалификация дистрибьюторов
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе — 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях — непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.

Получение UDI и регистрация в системе EUDAMED
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация — организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
Классификация изделий медицинского назначения
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Обучение: CE-маркировка и регуляторная система ЕС для медицинских изделий
Технические файлы и досье разработки
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований — в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
ISO 13485 и система менеджмента качества (СМК)
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.
Авторизованное представительство в ЕС (EC REP)
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Аудиты на соответствие требованиям Регламента ЕС 2017/745 и ISO 13485
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Взаимодействие с Нотифицированной Организацией
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Оценка и квалификация дистрибьюторов
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе — 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях — непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.
Ec rep что означает маркировка

1.Введение
Маркировка медицинских изделий должна играть важную роль при проектировании, разработке, производстве и остальных этапах жизненного цикла медицинских изделий, в том числе при эксплуатации. К сожалению, практика показывает, что производители либо не уделяют ей и её описанию должного внимания, либо весьма ловко и искусно скрывают свои усилия по этим направлениям. Например, почему в руководстве по эксплуатации нет или очень мало информации о маркировке, её содержании, расположении? Или почему при изучении стадий проектирования или производства медицинского изделия того или иного производителя достаточно часто нет информации о маркировке изделия? Или при изучении файла менеджмента риска медицинского изделия –часто из файла удается только понять, что маркировка на изделии есть, а речи об опасностях и рисках того или иного рода, связанного с маркировкой медицинского изделия, её эксплуатационной пригодностью и «читаемости» ничего не указано?
Конечно же, приведенное выше мнение может показаться чрезмерно пессимистичным. Попробуем разобраться, для чего нужна маркировка, какую информацию о ней требуется или желательно указывать. Причем начнем не с «тяжелой артиллерии» — требований, постановлений, стандартов, а с «конца» — то есть с того, что обычный пользователь изделия, не обязательно медицинского, привык видеть каждый день на маркировках и сопроводительной документации.
2.Идентификация изделий
Можно предположить, что обычный пользователь ничего не знает о требованиях к маркировкам, их содержанию, что их регламентирует, как нужно их описывать. Это в большинстве случаев верно – но в то же время он может наблюдать их повседневной жизни по много раз в день, и таким образом у него складывается свое собственное мнение, что же там должно быть указано. И «поймав» обычного пользователя с предложением пройти опрос, что должно быть на маркировках тех или иных продуктов, можно оказаться достаточно сильно удивленным, когда оный пользователь перескажет Вам чуть ли не большую часть требований стандартов или регламентов, не имея вообще никаких знаний, кроме тех, что такие требования в наше время где-то должны быть описаны. Но эти знания он «приобрел», так как он видел одну и ту же информацию, вернее, один и тот же набор приводимой информации плюс-минус, много тысяч раз.
Можно достаточно долго развивать эту мысль, приводить примеры того, что «клиентоориентированность», «эксплуатационная пригодность» и прочие явные и не явные параметры и свойства важны. Причем весьма вероятен и такой результат изучения определенного продукта пользователем – «у этого производителя это указано / написано / предусмотрено, а у этого – нет, значит первый лучше при прочих равных», то есть можно поговорить и о конкурентной способности изделий. Но вернемся все же к конкретике — информации на маркировках. Приведем ниже предположительное описание информации, полученной от «обычного пользователя» о том, что должно быть на маркировках, с некоторыми комментариями и примерами.
Итак, что же обычный пользователь привык видеть в первую очередь на маркировках изделий? Это наименование продукта – то есть верная и корректная идентификация изделия, скажем так, это аналог ФИО в паспорте в общем смысле этого слова. Иначе, это ответ на вопрос «что находится перед нами?».
Далее — во всех ли случаях указания наименования будет достаточно? Или, скорее, что должно включать в себя наименование? Предположим, что производитель делает несколько моделей под одним наименованием – получается, что наименование должно включать в себя и уточнения, если такие имеются. Большинство производителей, конечно же, так и делает – указывает наименование и модель. Или указывает наименование и номер по каталогу – REF номер (или его аналог). Или все три параметра. Но тут вернемся к предыдущему абзацу и примеру из него – «ФИО в паспорте». Можно было обратить внимание, что это не очень удачный пример – так как в может быть много человек с одним и тем же ФИО в одной стране. И тут-то как раз уже можно выводить достаточно много производителей на «чистую воду» — словосочетание «верная и корректная идентификация изделия» несет в себе смыслы и «полная», и «окончательная» идентификация. Приведем несколько примеров – производитель выпускает свои изделия в разных цветах / разных оформлениях, цвет (оформление) не указан ни в модели, ни в номере по каталогу, ни где-либо еще. Производитель подумает – а зачем? Не будем предполагать, что подумает пользователь о производителе, если он стоит на складе перед двумя одинаковыми коробками без возможности определить, какая коробка содержит изделие нужного ему цвета (оформления).
Можно сказать, что цвет не имеет влияния на функциональные характеристики – не будем заострять внимание на изделиях для детей или для взрослых. Приведем другой пример – производитель поставляет под видом одной модели изделия с разным внутренним устройством – то есть с разными функциональными характеристиками и, более того, более широким-узким назначением. Здесь уже пользователь не во всех случаях по внешнему виду, как для цвета (оформления), сможет понять, какая модификация где. И это очень печально.
Таким образом, подведем итог рассуждениям по поводу наименования, оно же – «верная и корректная идентификация». Пользователь при изучении «полного наименования» изделия, в том числе на маркировке, которое может включать: наименование изделия, модель и номер по каталогу или их аналоги, доп. параметры – не должен задумываться «а правильно ли я все понял», искать какие-либо отличительные признаки, присутствующие на одной модели, но отсутствующие на другой и так далее – информация для идентификации должна быть достаточной и исчерпывающей. Особенно это важно при ситуациях, требующих «экстренных» мер – любая ошибка или задержка в таких случаях может привести к нанесению вреда или чему-либо похуже.
Теперь расширим наш термин «идентификации» другими параметрами — это информация о производителе изделия, его адресе и контактных данных, например, телефоне, адресе электронной почты, сайте – по отдельности и вместе. По сути, это не только вопрос идентификации изделия, так как вполне возможен, пусть даже и с небольшим шансом, случай, когда изделия будут названы полностью одинаково (или производители будут названы одинаково, но располагаться в разных странах), но и указание информации о главном «ответственном» за данное изделие лице. Также для пользователя этот параметр важен, так как чем более известен производитель, там больше доверия он вызывает при прочих равных, тем более качественным считается изделие. Статистически, здесь проблем не бывает – производитель всегда указывает информацию о себе. Проблемы возникают в другом – в актуальности этой информации при изменении её частей – наименовании производителя, его организационно-правовой формы, адреса и т.д. Мы не заостряли внимание на этом раньше – но любая информация, сопровождающая изделие, в том числе на маркировке, должна быть актуальной. И её обновление должно происходить максимально быстро.
Дополнительный параметр, который вытекает из указания информации о производителе – это место производства изделия. Если у производителя несколько производственных площадок или адрес расположения производственной площадки отличается от места расположения офиса производителя, то следует указать и эту информацию, опять же, утрированно, в рамках идентификации и вопроса «кто виноват и где его искать?». Для пользователя этот параметр важен, опять же, по вопросам доверия – чем более «надежной» считается страна расположения производственной площадки, тем большое доверие получает изделие.
Вполне резонно заметить, что если производитель находится за рубежом, то ему необходимо иметь некоторое региональное представительство, как минимум, по законодательным причинам и наличию языковых барьеров, и указывать на маркировке информацию об его Уполномоченном представителе на некоторой территории (некоторой стране, группе стран). Примером данного требования является хорошо известный «EC REP» — Уполномоченный представитель в Европейском сообществе, символ и информацию о котором с 100% вероятностью можно увидеть на маркировках медицинских изделий в ЕС (если он сам не располагается на территории ЕС или его обязанности делегированы).
Укажем ещё один очевидный параметр, который требуется не забыть привести на маркировке – серийный номер или номер партии. Он указывается для идентификации или отслеживания конкретного экземпляра или конкретной партии изделий. Например, для определения, какой конкретно экземпляр изделия требуется отправить на плановое обслуживание, если их несколько на балансе. Или в серийном номере или номере партии достаточно редко, но иногда может кодироваться информация о производственной площадке или дате производства, или модели, или проч. Не забудем и про даты – вот, на что именно пользователь обращает внимание в одну из первых очередей на маркировке и упаковке изделия – дата производства, и, если применимо – дата истечения срока годности (или аналоги) – о них подробней поговорим чуть позже.
Рассмотрим частный случай – изделие представляет собой многоэлементное – чаще в случаях многоразового изделия. Тогда очень важно не забыть промаркировать каждый (при возможности) элемент изделия, и на маркировке элемента указать, как именно он называется. Иначе применение изделия будет затруднено, так как пользователю в общем случае может потребоваться больше времени на изучение, что это за элемент, как его применять, куда его присоединять и т.д.
И еще один частный случай, который скорее относится к дополнительной идентификации производителя, линейки продуктов или конкретного изделия – это товарные знаки. На маркировках часто располагают зарегистрированные соответствующим образом и находящиеся под охраной на определенной территории товарные знаки. Пользователь очень часто наблюдает их на маркировке или упаковке. И, опять же, бренд или товарных знак может вызывать у него разный уровень доверия к изделию. Но достаточно небольшому количеству пользователей будет понятно, почему мы привели именно такую формулировку ранее — «зарегистрированные соответствующим образом и находящиеся под охраной на определенной территории…». Очень часто случается, что производитель забывает, что товарный знак должен быть защищен на территории страны применения его изделия, иначе требуется убрать знаки охраны товарных знаков, информацию о защите товарного знака и проч.
Просуммируем вышеприведенное про информацию на маркировке изделия в рамках идентификации и что на ней требуется обычно указывать:
- Наименование изделия – обязательно для идентификации.
- Модель, номер по каталогу и проч. — обязательно (при наличии) для идентификации.
- Информация о производителе – обязательно для идентификации.
- Производственная площадка – обязательно (при наличии) для идентификации.
- Информация об уполномоченном представителе – обязательно или рекомендуется для идентификации лица, ответственного по определенным активностям на определенной территории обращения изделия.
- Серийный номер или номер партии, даты – обязательно для идентификации.
- Наименование элемента изделия – обязательно для идентификации элемента многоэлементных и, как правило, многоразовых изделий.
- Товарный знак – по желанию при наличии надлежащей регистрации в стране применения.
3.Применение изделия
Перейдем к вопросу – «что обычно располагается на маркировке изделия в целях его надлежащего применения?».
Начнем с того, что на большинстве маркировок располагается указание на применение в соответствии с сопроводительной документацией – то есть на руководство по эксплуатации (инструкцию по применению, руководство пользователя и проч.). Это делается для того, чтобы напомнить пользователю, что изделие допускается применять в соответствии с содержанием сопроводительной документации, и ознакомление с ней перед использованием – обязательно абсолютно для всех изделий (изучение сопроводительной документации и / или описания на упаковке / изделии), и никак иначе. Шутки типа: «инструкцию читают только тогда, когда нечего читать, или уже все безнадежно сломано» — крайне несмешные, особенно для изделий повышенного риска для здоровья, таких, как медицинские изделия. Приводите пользователю требование об обязательном изучении сопроводительной документации везде, где только можно и разумно – пользователь крайне не любит читать инструкции от слова совсем.
Далее, на маркировке изделий также располагают важные запретительные / разрешительные знаки – содержит тот или иной материал – для пользователей с аллергией, использовать повторно нельзя, использовать можно только в помещении, использовать совместно только с таким изделием т.д.
Здесь необходимо заострить внимание на одной проблеме, которая встречается пугающе часто – отсутствие должного описания маркировки и её содержания, расположения. Пользователь, например, может сказать, в некоторых случаях весьма резонно, – «я вижу символ, но не вижу его описания — как я могу понять, что он означает?». Любая ситуация, которая может вызвать вопросы у пользователя – это удар по безопасности и эффективности изделия. Это шанс на возникновение ненадлежащего применения изделия. Практически никогда нет явных причин не указывать описание содержания маркировки.
Еще одним важным параметром, который указывают на маркировке – это даты и временные промежутки – даты изготовления, срок годности, дата окончания срока годности и проч. в различных комбинациях. Для определенных изделий (чаще многоразовых) – это в основном дата изготовления. Для других типов изделий, когда критически важно привести срок годности изделия именно на маркировке (одноразовые стерильные, например), основной акцент приходится на дату окончания срока годности – после её наступления строго запрещено применение изделия. Вариантов указания дат и сроков на маркировке много – только дата изготовления (если в сопроводительной документации указан срок эксплуатации), или дата изготовления и дата окончания срока годности, или дата изготовления и срок годности и др. Главное – точная и простая идентификация даты изготовления и даты окончания возможного применения медицинского изделия.
Также немаловажными параметрами, которые требуются указать на маркировке – это характеристики изделия – их набор зависит от конкретного типа изделия. Например, это могут быть размеры рабочей части – их указывают не «для галочки», а чтобы дать возможность пользователю быстро разобраться, подходит ли данное изделие для применения в конкретном случае. Или электрические параметры – самое простое объяснения их наличия: можно или нельзя подключить в данном месте к сети питания.
Перейдем к очень важному вопросу, который задается производителями очень часто, но вызывает искреннее недоумение – «нужно ли приводить маркировки (и сопроводительную документацию) на языках места обращения изделия?». Обычного пользователя такой вопрос, скорее всего, также сильно удивит. Он спросит: «а как мне применять изделие, если я знаю только родной язык?». Как можно говорить о безопасности и эффективности применения изделия, если пользователь не понимает, что написано на маркировке? Более того, предположим, что мы спросим профессора-лингвиста в каком-нибудь университете: «Скажите, пожалуйста, как Вы думаете, Вы лучше понимаете язык, который изучаете почти всю свою жизнь, по сравнению с обычным носителем языка?». Скорее всего, уважающий себя профессор ответит, что он был бы не столь уверен, что «осилит» «носителя языка» в этом вопросе – то есть подтверждения «полного понимания» написанного на иностранном языке у пользователя, пусть даже и с некоторым приличным уровнем «владения языком», нет. Из чего можно сделать вывод – есть ли требования к указанию маркировок и их содержанию или нет на родном языке в области обращения – не важно, их нужно указывать на языке области обращения (или приводить мультиязычную / несколько моноязычных маркировок).
4.Выводы
Подведем итог наших размышлений – что должно быть приведено и как должно быть приведено на маркировке.
Маркировка должна или может содержать:
- Наименование изделия
- Модель, номер по каталогу и проч.
- Информация о производителе.
- Производственная площадка.
- Информация об уполномоченном представителе.
- Серийный номер или номер партии
- Даты изготовления или даты изготовления и срок окончания применения.
- Наименование элемента изделия.
- Товарный знак.
- Ссылку на «использовать в соответствии с сопроводительной документацией».
- Разрешающие или запрещающие знаки по надлежащему применению изделия.
- Соответствующие данному типу изделий параметры и характеристики.
И, что очень важно, содержать информацию на языке области обращения изделия.
Ec rep что означает маркировка
Определение Директив ЕС, требованиям которых должна соответствовать продукция:
93/42/ЕЭС – для медицинских изделий (MDD) or 90/385/ЕЭС — для активных имплантируемых медицинских изделий (AIMDD).
Определение классификации продукции с помощью приложения IX Директиву для медицинских изделий (MDD):
Класс I (нестерильные, без фунции измерения),Класс I (стерильные, с фунцией измерения),Класс IIa,Класс IIb или Класс III/AIMD*.
* Активные имплантируемые медицинские изделия подлежат тем же нормативными требованиями как изделия Класса III
Для всех устройств, кроме Класса I (нестерильные, без функции измерения), осуществить Систему Менеджмента Качества (СМК),
в соответствии с Приложением II или Приложением V директивы MDD.
Для продуктов Класса III/AIMD, подготовить Дизайн Досье (Design Dossier*).
Для всех остальных устройств, подготовить ЕС Технический файл, который содержит подробную информацию подтверждающую соответствие медицинского изделия
директиве MDD 93/42/ЕЭС.
* Для устройств Класса III / AIMD будут необходимыми данные клинического исследования. Клинические испытания в Европе должны быть предварительно одобрены европейским нотифицированным органом.
Назначить Европейского Авторизованного Представителя (EC REP), который расположенный в Европе.
Поместить название и адрес ЕС REP в инструкции по применению и на упаковке.
Авторизованный
Представителт
Для всех устройств, кроме Класса I (нестерильные, без функции измерения),
Ваша СМК и Технический Файл или Дизайн Досье должны пройти проверку Нотифицированным Органом, который является третьей стороной, аккредитованной Европейскими властями для оценки медицинских изделий и компаний — производящих продукты медицинского назначения.
Подготовка Декларации Соответствия, которая является документом имеющим обязательную юридическую силу, подготовленным изготовителем с указанием,
что устройство соотвечает приенимым Директивам.
Для всех медицинских изделий и средств IVD в Европейском Союзе, сертификат CE является необходим. Эта сертификация подтверждает, что устройство соответствует всем нормативным требованиям «Медицинских Директив».
Choosing an EU Authorised Representative (EC REP) for Medical Device Regulatory Issues
%20For%20Medical%20Device%20Regulatory%20Issues.png)
So you’re ready to export to Europe. You’ve tackled the myriad tasks associated with packaging, marketing, shipping, and distributing your medical devices. But wait, a host of regulatory issues still loom.
One such issue that confronts all non-European manufacturers that sell devices in Europe is the appointment of an Authorised Representative . In their zeal to get products out the door, most people don’t give it much thought, but there are good reasons why you should.
What is an Authorised Representative (EC REP)?
Essentially, an EC REP plays an intrinsic part in post-market surveillance, including vigilance. The Authorised Representative (AR) is your link to European authorities, and they must maintain physical presence in Europe. They will register your medical device or IVD before it is marketed, and will always be available to serve as a contact between you and the Competent Authorities of the EU member states.
Your AR will also have access to your Technical File(s), which must be available for inspection by the Competent Authorities. In other words, a variety of the responsibilities of the manufacturer are delegated to the Authorised Representative. The name and address of your AR must be placed on the information that accompanies your device, such as (packaging) labeling and instructions for use.
In the event of an incident, your AR will assist and coordinate the reporting of the incident to the Competent Authorities, and will cooperate with you and your distributors to ensure that the proper reporting and follow up protocols are followed. In the event that your device is withdrawn from the market, your medical device Authorised Representative can represent you to the European Commission for consultation.
These are the tasks that you delegate to your representative to perform on your behalf. However, it is important to note that the EC REP is not legally responsible for non-conforming devices, unless the problem is a direct result of one of these tasks.
The manufacturer should always responsible for the safety of the device. Some of the Member States’ Competent Authorities however are of the opinion that the Authorised Representative can be held responsible for the device. This opinion provides a challenge for the Authorised Representative which has no control over design, manufacture, packaging and labeling of a device, otherwise the AR would be a manufacturer.
Choosing an European Authorised Representative
Make sure you know exactly what the role is of the AR and which of the responsibilities you need to delegate. At minimum, an AR must be able to handle (near) incident reporting, product recalls, complaint handling, and post-market feedback.
Each AR provides slightly different services, although many will custom tailor a contract for you, and obviously fees will vary. Determine which EC REP services you need, and how much you are willing to pay, and choose an EC REP who most effectively (and efficiently) can meet those needs.
Regardless of your product, you should look for an AR who has considerable European regulatory experience with medical devices or IVDs. European regulations and vigilance can be complicated, and your AR should have experience with a wide range of devices. When talking to different AR providers, your Authorised Representative need not always have to have the expert knowledge of your device and technology, but they should be able to guide you through the regulations and requirements that apply to your device.
Considering the role of the AR, it is important to ensure that the responsibilities of both you, as the manufacturer, and your selected AR are clearly outlined and stipulated in a contract.
Using a distributor as your Authorised Representative in Europe
Some companies opt to use their distributor or importer as their Authorised Representative. Many distributors, however, may not be aware of the role and responsibilities of Authorised Representation. There are other issues to consider.
Your distributor’s name and address will be on all of your materials, no matter where they are sold in Europe. If you decide to change distributors, you will have to reprint all of your labels, manuals, packaging, and are presented with a potential challenge on how to deal with the products on the market that have that distributor’s name on them. Besides, if you choose one distributor among several to be your Authorised Representative, such an arrangement may create ill will among your other distributors.
A distributor should focus on the sale and marketing of your devices, not on regulatory affairs. If the European laws and guidelines are modified, are you sure your distributor will keep abreast of these changes and notify you when changes affect your devices? In the regulatory affairs world, ignorance is not bliss.
Your distributor, as your Authorised Representative, will have access to your Technical File(s), which may include proprietary information. A related issue deals with recall and (near) incident reporting. If the Competent Authorities question an incident or a noncompliance that occurred with your product, can you be sure your distributor will defend your company. or protect their own interests? That being said, companies that sell Class I devices who only intend to sell in a few countries through a single distributor might try to save some money appointing the distributor as their AR. However, since the annual cost of appointing an AR is generally under US$3,500, you may want to weigh the risks versus benefits of such a decision.
Regardless of whether you decide to choose a distributor or an independent party as your AR, you should select someone who has some history in the business. If the company you select is only a few years old, they may end up going out of business.
In that case, guess who pays the bill for reprinting Instructions for Use, labels and packaging? Furthermore, a company with more history and experience in the business may have stronger personal relationships with Competent Authorities, which may work to your advantage if problems arise.
Increased scrutiny from European authorities
European officials will continue to tighten regulations regarding vigilance. Remember that the manufacturer must ensure that their devices conform to applicable standards and directives — neither your AR nor a distributor can guarantee this for you.
Therefore, it is important to choose an Authorised Representative who has strong connections in Europe, who stays on top of changes in regulatory affairs, and who makes serving your representation needs their first priority.
Monitoring and complying with European regulations can be challenging. Choosing an Authorised Representative for your medical device or IVD company is a serious decision, one which will affect you for years. Careful consideration of the points outlined above — your needs, their experience, the company’s history, and the question of appointing a distributor — will help you make the best decision for your company.
This content was originally published by EMERGO by UL and can be viewed by clicking here.